Create color-coded SNP BED from VCF
A color-coded variant file where SNPs from each allele are a different color and the homozygous SNPs are also a different color improves visualization.
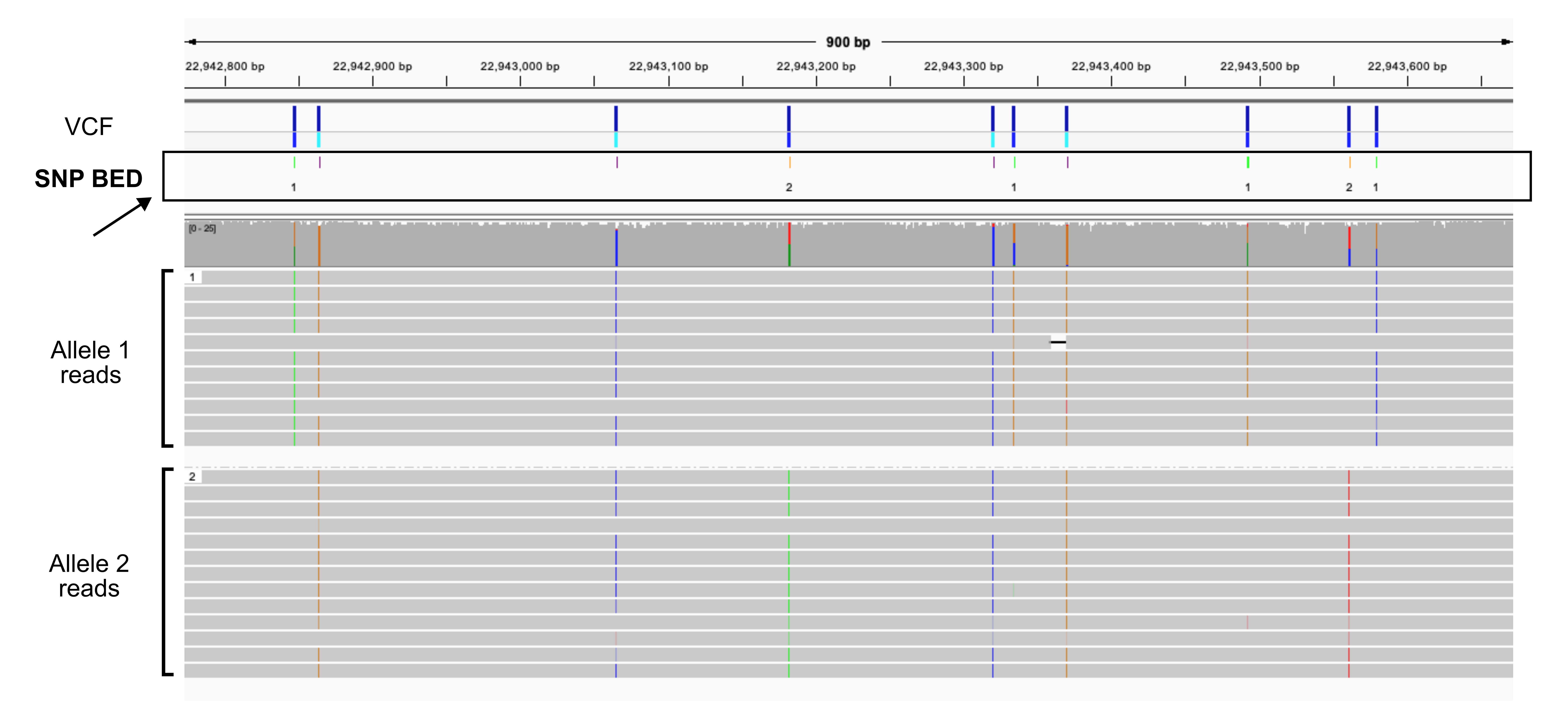
Create the BED files
The script that generates this BED file works on a VCF file that is only single-sample (like from de novo variant calling), phased (separated by haplotype), and diploid (only two alleles).
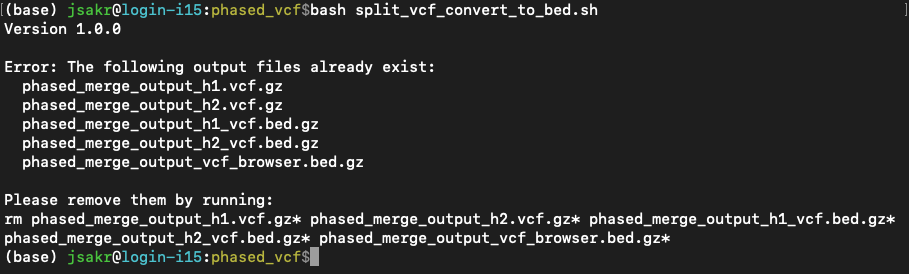
When you run the script, it will first check if any of the 5 output files already exist. If they do, it stops and prints an error message along with a rm command for you to copy and paste into your terminal. This is to prevent you from accidentally overwriting previous files.
The error message will look something like this:
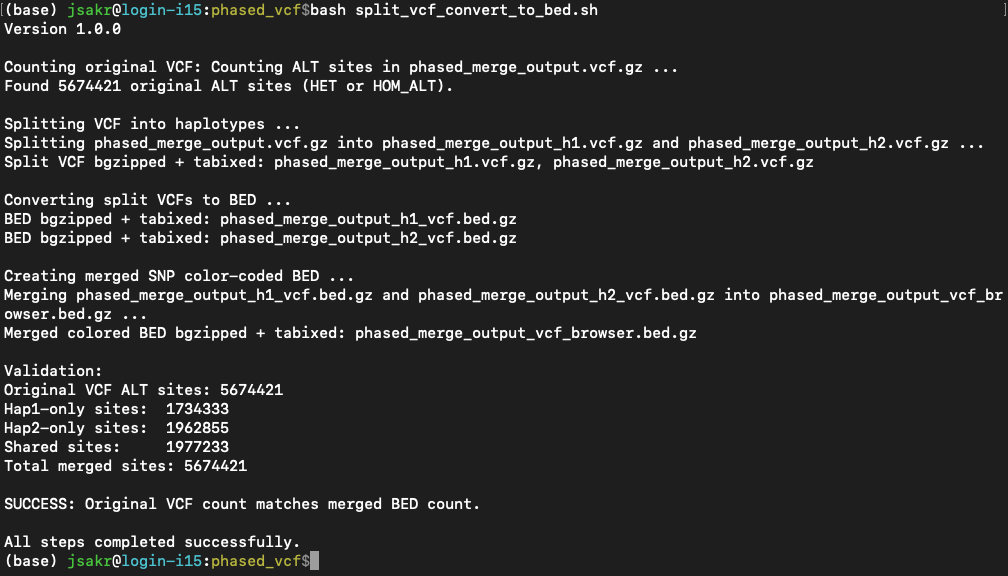
Then it will run through the following steps:
- Step 1: Split VCF: It splits the input VCF into two new VCF files, one for each allele, and index them.
- Step 2: Convert to BED: It takes the two new VCF files and converts them into BED files.
- Step 3: Merge BED and Color: It takes the two BED files and merges them into a single, final BED file where the SNPs are color-coded.
- Step 4: Validation: Compares the number of variant sites found in the original input VCF file to the total number of sites in the final merged BED file.
The output in the terminal will look something like this:
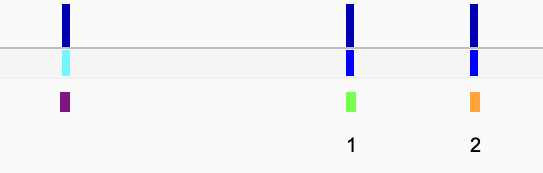
Currently, the color correspond to Allele 1 = green, Allele 2 = orange and homozygous = purple. Only the allele-specific SNPs are labelled (you can pick colors you want or the labels by editing the script).
The following output files will be created based on the input basename:
<prefix>_h1.vcf.gz<prefix>_h2.vcf.gz<prefix>_h1_vcf.bed.gz<prefix>_h2_vcf.bed.gz<prefix>_vcf_browser.bed.gz(and all corresponding index files)
The Script
You must have cyvcf2, pandas, bgzip, and tabix installed in your environment.
You can create an environment using conda by running:
conda create -n vcf_env -c bioconda -c conda-forge cyvcf2 pandas htslib
Here are the versions that work with this script (at time writing this):
| Package | Version |
|---|---|
| cyvcf2 | 0.11.7 |
| pandas | 1.3.5 |
| tabix (htslib) | 1.20 |
| bgzip (htslib) | 1.20 |
To run the Python code, use a Bash wrapper script that looks something like this:
# activate env with necessary packages
conda activate vcf_env
# run the script and point to the VCF file
python split_vcf_convert_to_bed.py phased_merge_output.vcf.gz
Get the python code in split_vcf_convert_to_bed.py here.